


Description
High-performance computing
Computational modeling is a priority tool for solving scientific and engineering problems in physics, engineering, biology, generating new principles for conducting research and applications.
The Centre for Advanced Research is conducting studies into the precise atomistic simulation of substances aimed at finding new ideas and ways to achieve the advanced level of work in the development of new biologically active substances and functionally different materials for different purposes, such as:
- The use of existing and development of new methods of modeling of nano- and biosystems;
- Prediction of biological activity of the low molecular weight substances, carbon nano-clusters, interacting with each other and proteins, nucleic acids, polysaccharides, membranes and heterogeneous ensembles of biomolecules;
- Predictive assessment of biological risks associated with the use of nano-particles of different nature;
- Nanobionics and atomic-molecular design nanobiomimetical systems and materials;
- Molecular engineering of new components for materials separation and purification, nanotechnology and optoelectronic devices;
- New active/
New materials
Modern methods of computer simulations allow us to predict many of the properties of developed materials. There are cases where the application of computational testing reduces the cost of development by thousands times. Pilot design of new products with given properties and technologies of their production is becoming standard for theof industry materials. Powerful enough clusters are able to "browse" and within weeks "test" commercial catalogs, including hundreds of thousands of substances. Unfortunately, searching for composite materials consisting of different substances, the number of combinations requiring sorting, is inconceivable.
Modeling of drugs
Powerful PC and especially clusters allow calculations of possible medicine candidates, that interact with individual proteins or other functionally important biomolecules. Atomistically aquired accurate data results are very important, but the duration of the computational experiment is often comparable with the time of the actual test.
The introduction of supercomputers significantly modifies the possibilities of search and design of drugs:
- Firstly, there is the possibility of advancing simulation of entire "libraries", i.e. hundreds of thousands of substances that can interact with a selected biological target,which critically reduces the volume of excess chemical synthesis and biological screening of candidate substances on the role of drugs.
- Secondly, for selected candidates it is possible to predict interactions with hundreds of different targets. There is a possibility of mapping the expected activities of the candidate, which qualitatively changes the procedures for identifying promising areas of the use of drugs in the treatment of various diseases and a priori estimates possible biological risks.
- Thirdly, previously it was not possible, but now we are able to build accurate models of atomistically drug interactions with a variety of carriers and those ensembles of complex biomolecules that determine the processes of absorption from the injection place, transportation to a variety of tissues of chemical biotransformation ofexcretion of developted substances.
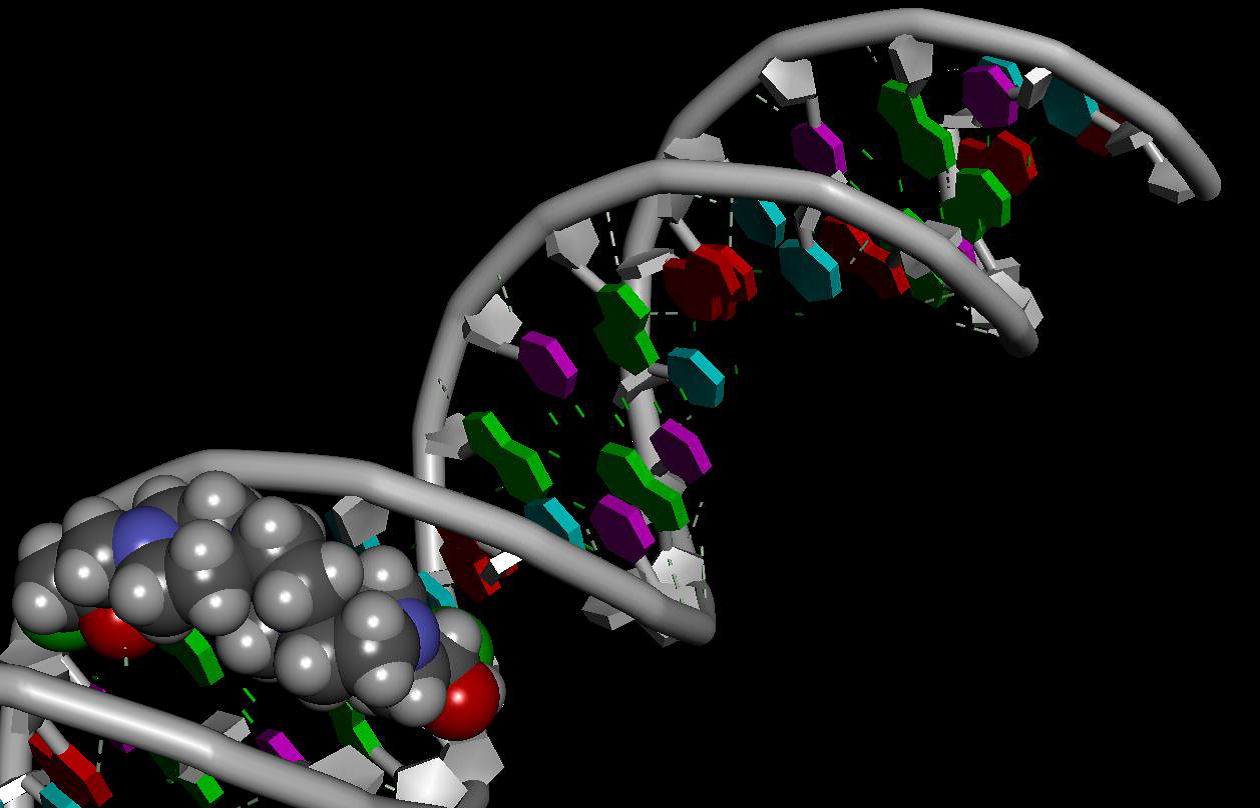
Domestic anti-cancer drug that interacts with its target - the DNA molecule